Was sind Zilien?
Zilien sind haarähnliche Zellfortsätze, die auf beinahe allen menschlichen Zellen zu finden sind. Man unterscheidet bewegliche Zilien (motile Zilien) und unbewegliche (immotile) Zilien, die auch als Primärzilien bezeichnet werden. Während Primärzilien eine Organelle fast jeder Zelle darstellen, finden sich motile Zilien nur auf bestimmten Epithelien (z.B. Luftwegsepithel) im Verbund von mehreren Tausenden (Cluster). Die Hauptaufgabe der motilen Zilien besteht darin, einen gerichteten Transport von z.B. Flüssigkeiten im Körper zu erzeugen.
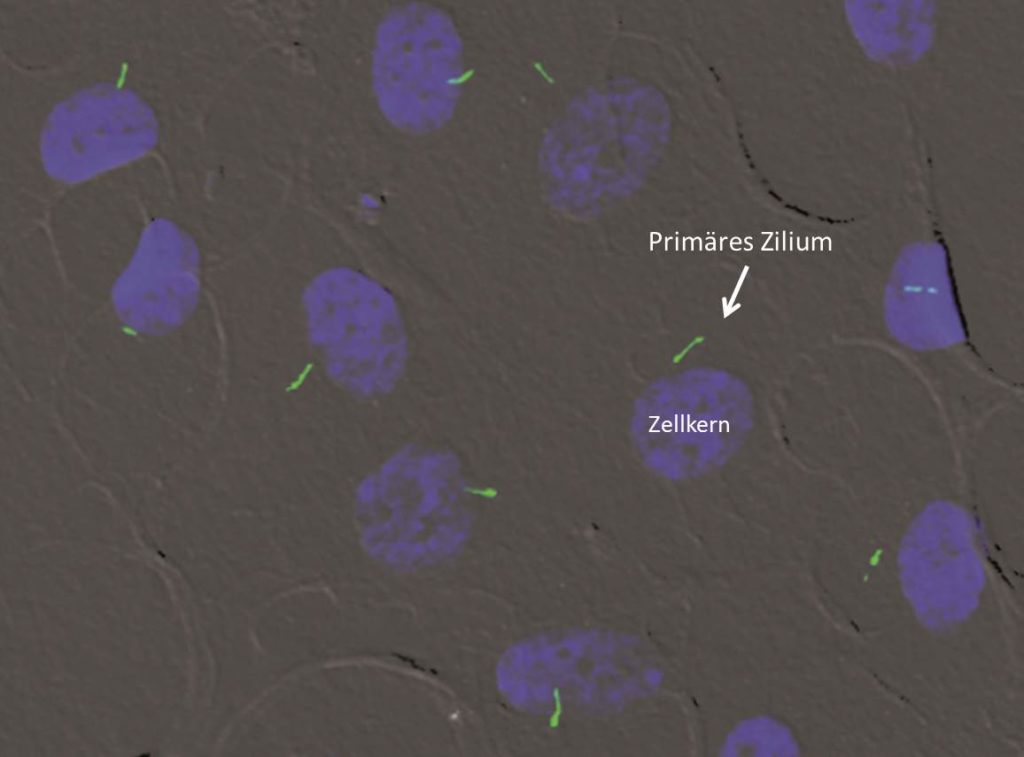
Die Funktionen von Primärzilien hingegen sind vielfältig. Sie bestehen im Wesentlichen in der Übertragung äußerer Reize an das Zellinnere. Bei diesen Reizen kann es sich um Licht, Gerüche, Fluss, Osmolaritätsgradienten oder anderweitige Reize handeln. Primärzilien stellen damit auch funktionell eine Art Antenne der Zelle dar, die für eine Verbindung zwischen äußerem Milieu und Zellinnerem sorgt.
Obwohl Zilien als Zellorganelle bereits 1800 beschrieben wurden, blieb ihre Bedeutung für lange Zeit ungeklärt. Erst 1979 konnte ein Zusammenhang zwischen der Funktion beweglicher Zilien und einer chronischen Lungenerkrankung mit gleichzeitiger Störung der Organlateralisation (primäre Ziliendyskinesie/Kartagener-Syndrom) hergestellt werden. Als Ende der 90er Jahre darüber hinaus ein Zusammenhang zwischen der Entstehung zystischer Nierenerkrankungen und Störungen der Zilienfunktion offensichtlich wurde, erlebte das Interesse an Zilien und deren Erforschung eine ungeahnte Renaissance. Heute weiß man, dass Primärzilien eine zentrale Rolle für diverse zentrale biologische Prozesse darstellen, u.a. für die Detektion verschiedenster sensorischer Reize, für die Zellteilung, die Embryogenese oder Zellreparatur-Mechanismen. Hierbei kommt es zur Aktivierung zahlreicher intrazellulärer Signalwege, wobei die exakten Abläufe weiterhin ungeklärt und Gegenstand aktueller Forschung sind. Erkrankungen, deren Ursache in einer gestörten Zilienfunktion liegt, werden daher heute unter dem Begriff „Ziliopathien“ zusammengefasst.
Zilienaufbau
Die Zilienarchitektur besteht aus 9 zirkulär angeordneten Mikrotubuluspaaren, die im Falle motiler Zilien um ein zentrales Tubuluspaar angeordnet sind (sog. 9+2 Struktur). Im Falle immotiler Zilien fehlt das zentrale Tubulus-Paar.
Anhand der Architektur sowie der Funktion unterscheidet man drei Zilienformen:
| 1) Bewegliche Zilien haben eine „9×2 + 2 – Struktur“ und verursachen in großen Verbänden einen gerichteten Fluss. Innere und äußere Dyneinarme sorgen durch aneinander vorbeigleiten der Mikrotubuli für die gerichtete Bewegung im Zilium. Bewegliche Zilien findet man vor allem im Respirationstrakt, wo sie Mukus aus dem Körper transportieren. Außerdem in der Tuba uterina, wo sie den Transport der Eizelle aus dem Ovar in Richtung Uterus bewirken sowie auf cerebralen Ependymzellen, welche für einen gerichteten Liquorfluss sorgen. Das Flagellum des Spermiums stellt ebenfalls ein Zilium dar, allerdings mit einer 9+1 Struktur. | 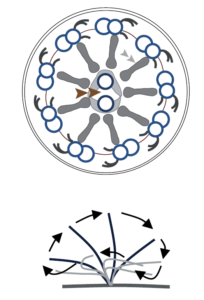 |
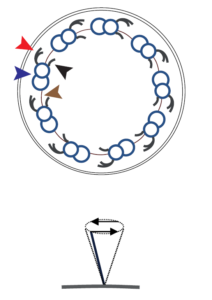
| 2) Ein weiteres bewegliches, aber solitär stehendes Zilium, ist das Zilium am sog. Embryonalknoten, einer entscheidenden Struktur der frühen Embryogenese. Dieses Zilium besteht aus ebenfalls 9 zirkulär angeordneten Mikrotubuluspaaren, die mit Hilfe von Dyneinarmen beweglich sind, allerdings fehlt das zentrale Tubuluspaar („9 x 2 + 0 – Struktur“). Hieraus resultiert eine rotierende Bewegung des Ziliums, die entscheidende Bedeutung für die korrekte Organlateralisation und damit die Ausbildung einer Rechts-Links-Achse des sich entwickelnden Emryos hat.
Eine Dysfunktion dieses Ziliums verursacht Erkrankungen aus dem Spektrum der Heterotaxien oder gar einen vollständigen Situs inversus.
|
 |
| 3) Unbewegliche Zilien (Primär-Zilien) bestehen aus 9 zirkulär angeordneten Tubuluspaaren ohne zentrales Tubuluspaar und ohne Dyneinarme (9x 2 + 0 – Struktur). Sie sind auf beinahe allen Körperzellen solitär zu finden. Nach heutigem Wissensstand haben sie zentrale Bedeutung in der Signalrezeption äußerer Reize und der Übermittlung dieser an das Zellinnere. | 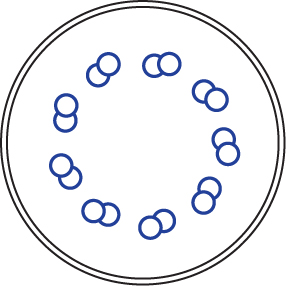 |
Entstehung der Zilien (Ziliogenese)
Zilien entstehen nur in bestimmten Abschnitten des Zellzyklus (G1, Quieszenz). Nach der Zellteilung entwickelt sich aus einer der beiden Zentriolen, der sog. Mutterzentriole, ein Basalkörperchen. Dieses wiederum ist Ausgangspunkt der Ziliogenese, d.h. aus dem Basalkörperchen heraus erwächst ein Zilium. Da Zilien selbst keine Proteine herstellen können, müssen alle notwendigen Bauteile der Ziliogenese in das Zilium transportiert werden. Die exakten Abläufe dieses Transports sind bislang noch nicht endgültig verstanden. Manche Zilienproteine werden mittels Vesikeln (Transportblasen) an die Zilienmembran transportiert und dort freigesetzt. Dies erfolgt über einen hochkomplexen Transport entlang des Zilien – Axomers, bei dem sowohl Proteine vom Zytoplasma zur Zilienspitze als auch umgekehrt transportiert werden. Dieses Transportsystem wird als intraflagellärer Transport (IFT) bezeichnet. Genetische Defekte, die zu Störungen des IFTs oder seiner Bestandteile führen, wurden als Ursache diverser Ziliopathien, u.a des Bardet – Biedl Syndroms sowie der Gruppe der Short – Rib – Polydaktylie Erkrankungen identifiziert.
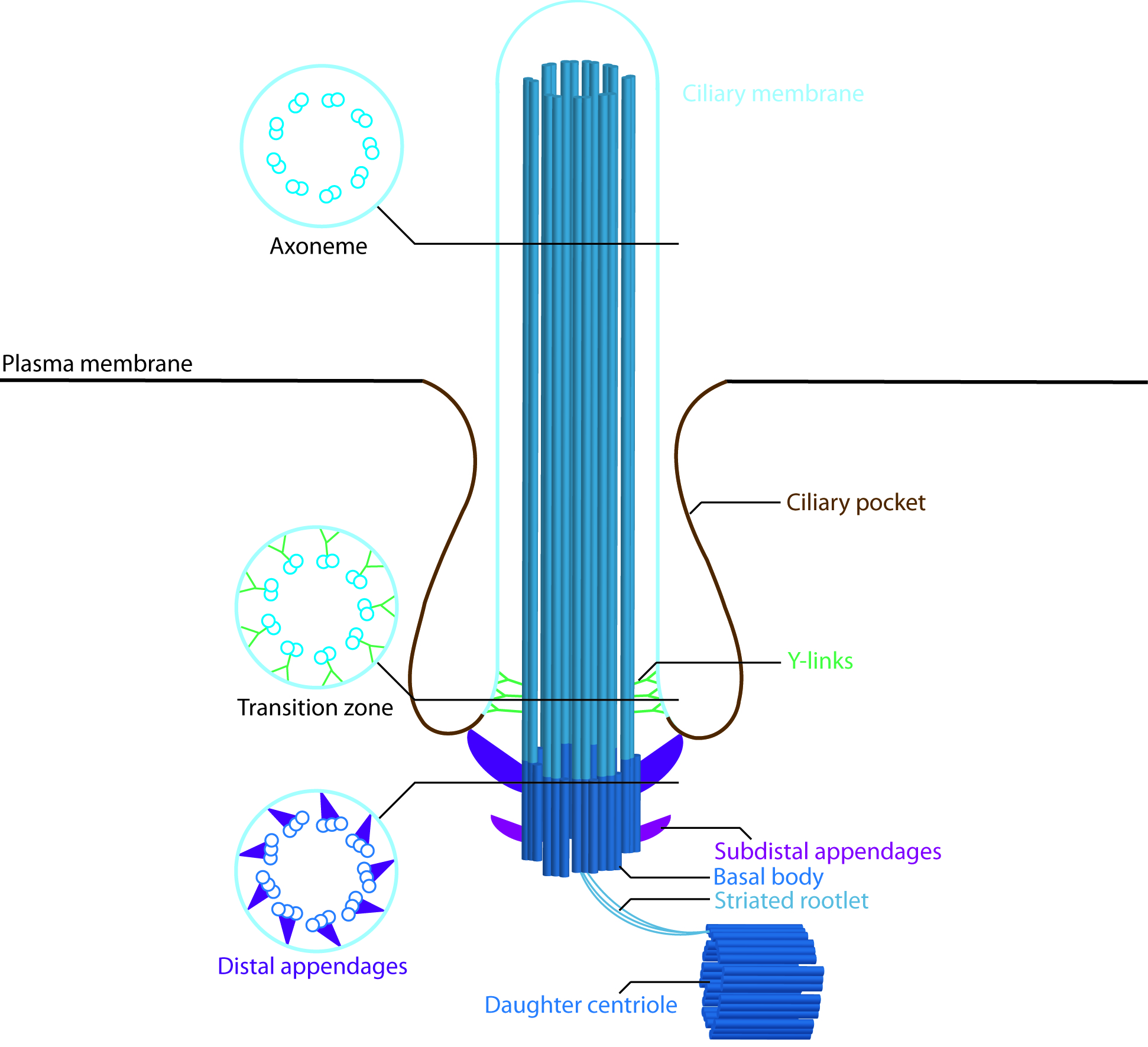
Zilienstruktur
Entlang des Ziliums unterscheidet man von Spitze zur Basis folgende Abschnitte: Zilienspitze, Zilienkörper, Zilienhals, Transitionszone, das „Inversin – Kompartiment“ sowie das Basalkörperchen.
Zilien sind von einer speziellen „Zilienmembran“ umgeben. Die Transitionszone bildet hierbei die Barriere zwischen Zelle und Zilium, über welche nur ein selektiver, streng kontrollierter Proteinaustausch stattfinden kann. Viele Nephronophthise–Gene kodieren Proteine, die an diesem Bereich der Zilien lokalisiert sind und vermeintlich entscheidende Bedeutung für die Kontrolle des Transports über die Transitionszone haben.
Molekulare ziliäre Funktionen
Zellzyklus: Die Ausbildung von Zilien erfolgt nur in bestimmten Abschnitten des Zellzyklus. Es wird daher angenommen, dass Zilien selbst Einfluss auf den Zellzyklus haben könnten. Zwar existieren mittlerweile mehrere Hinweise, die eine solche These unterstützen, die exakten Zusammenhänge zwischen Zilienfunktion und Zellzyklus sind jedoch weiter ungeklärt.
Intraflagellärer Transport (IFT): Zilien verfügen über ein komplexes Transportsystem, welches einen Proteintransport aus der Zelle ins Zilium und umgekehrt gewährleistet. Dieses Transportsystem entlang des Zilien – Axomers wird als intraflagellärer Transport (IFT) bezeichnet und besteht aus verschiedenen Proteinkomplexen. Während „Komplex B“ für den Transport von der Zilienbasis zur Zilienspitze verantwortlich ist, koordiniert „Komplex A“ den retrograden Proteintransport von der Zilienspitze zurück zur Zilienbasis. Wird im Tierversuch der intraflagelläre Transport künstlich ausgeschaltet, versterben die entsprechenden Tiere noch im Mutterleib, was die essentielle Bedeutung dieser ausgereiften Transportprozesse hervorhebt. Bei Menschen führen Mutationen von IFT – Proteinen zu Defekten der Knorpel – und Knochenausbildung, was sich klinisch zumeist als sog. Short – Rib – Hexadaktylie Syndrom (Jeune Syndrom, Mainzer – Saldino Syndrom u.a.) bemerkbar macht. Außerdem konnten IFT – Mutationen mit dem klinischen Bild eines Bardet – Biedl Syndroms in Verbindung gebracht werden.
Ziliäre Signalwege
Mindestens 5 verschiedene intrazelluläre Signalwege haben eine Bedeutung in der Pathogenese verschiedener Ziliopathien:
- Wnt – Signalweg: Bahnbrechende Studien konnten sowohl an Zellkulturen als auch an Zebrafischen zeigen, dass der hoch konservierte Wnt Signalweg durch tubulären Urinfluss beeinflusst wird. Kommt es aufgrund ziliärer Defekte zur Störung dieser Regulation, kann dies fatale Auswirkungen auf die Ausbildung einer apikal – basalen Zellausrichtung renaler Tubulusepithelien (Prinzip der planaren Zellpolarität) und damit auf die Ausbildung regelrechter tubulärer Röhrenstrukturen haben. Eine entsprechende Interaktion mit dem Wnt – Signalweg konnte für verschiedene ziliäre Proteine (NPHP3, NPHP4) gezeigt werden.
- Hedgehog – Signalweg: Der Hedgehog–Signalweg ist von entscheidender Bedeutung für die Knorpel – und Knochenentwicklung während der Fetalzeit. Defekte führen zu ziliären Chondrodysplasien mit unterschiedlich schweren Knochendeformitäten. Zusätzlich können kardiale Fehlbildungen, Lateralisationsdefekte sowie zystische Nierenerkrankungen beobachtet werden. Als Beispiele sind die asphyxierende Thoraxdysplasie (Jeune Syndrom), das Mainzer – Saldino Syndrom oder das Ellis van Creveld Syndrom zu nennen. Für Mutationen verschiedener ziliärer Proteine (NPHP6, NPHP7, IFT140…) konnte ein Zusammenhang mit dem Hedgehog–Signalweg nachgewiesen werden.
- mTor: Bei polyzystischen Nierenerkrankungen (ARPKD/ADPKD) ließ sich eine Aktivierung des mTOR – Signalwegs nachweisen. Eine medikamentöse Blockade dieses Signalwegs mittels Everolimus oder Sirolimus führte im Tierversuch zu einer signifikanten Hemmung des renalen Zystenwachstums. Die Ergebnisse klinischer Studien an Menschen waren jedoch eher ernüchternd. Inwieweit der mTOR – Signalweg daher ein mögliches Ziel zur Therapie zystischer Nierenerkrankungen darstellen kann, ist derzeit eher kritisch zu betrachten.
- Hippo – Signalweg: Im Falle von NPHP4 – und NPHP9 – Mutationen konnten Veränderungen des Hippo Signalwegs beobachtet werden, deren Bedeutung noch nicht abschließend geklärt ist.
- DNA – Damage Response: In jüngster Zeit konnte außerdem gezeigt werden, dass NPHP9, NPHP14 und NPHP15 entscheidenden Einfluss auf hoch konservierte Prozesse der DNA – Reparatur haben.
Weitere: Darüber hinaus wurden weitere Interaktionen ziliärer Proteine mit intrazellulären Regulationsmechanismen berichtet, u.a. der Regulation des intrazellulären Kalzium – Transports sowie des TGF-ß und anderer Signalwege. Die exakten Zusammenhänge sind bislang ungeklärt und Gegenstand aktueller Forschung.
Was sind Zysten?
Bei Nierenzysten handelt es sich um flüssigkeitsgefüllte Hohlräume im Nierengewebe, die in unterschiedlicher Anzahl und Größe auftreten können. Die Ursachen sowie die Bedeutung von Nierenzysten können stark variieren. Während einzelne, sog. solitäre Nierenzysten zumeist als Zufallsbefund im Rahmen von Routine – Ultraschalluntersuchungen auffallen und klinisch bedeutungslos sind, handelt es sich bei polyzystischen Nierenerkrankungen um erbliche Krankheiten, die mit der Ausbildung mehrerer Zysten in beiden Nieren einhergehen. Je nach zugrundeliegender Erkrankung können verschiedene Abschnitte des Nephrons an der Zystenbildung beteiligt sein. Während bei der autosomal dominanten polyzystischen Nierenerkrankung (ADPKD) Zysten in allen Nephronabschnitten zu finden sind, bleiben sie bei den meisten anderen zystischen Nierenerkrankungen (ARPKD, Nephronophthise, BBS) auf das Sammelrohr beschränkt. Sowohl die Zahl als auch die Größe der Nierenzysten kann im Laufe des Lebens zunehmen und so gesundes Nierengewebe verdrängen. Das Eintreten eines dialysepflichtigen Nierenversagens variiert jedoch je nach zugrundeliegender Erkrankung erheblich und ist nicht alleine von der Anzahl oder der Größe sich entwickelnder Zysten abhängig.
Was haben Zilien mit Zysten zu tun?
Die exakten Abläufe, die zur Ausbildung von Nierenzysten führen, sind bis heute nicht vollständig verstanden. Allerdings hat das pathophysiologische Verständnis in den vergangenen 20 Jahren insbesondere durch die Identifizierung der genetischen Ursachen zystischer Nierenerkrankungen (sog. Zystengene) enorm zugenommen. 1985 wurde mit der Identifizierung von PKD1 als verantwortlichem Gen für die autosomal dominante polyzystische Nierenerkrankung (ADPKD) ein erster Meilenstein im molekularbiologischen Verständnis zystischer Nierenerkrankungen gelegt. Seither wurden annährend 100 Gene identifiziert, die in mutierter Form zur Ausbildung von Nierenzysten führen können. Ende der 90er Jahre des letzten Jahrhunderts konnte darüber hinaus erstmals an einem Fadenwurm (Caenorhabditis elegans) nachgewiesen werden, dass das von PKD1 kodierte Genprodukt (Polyzystin-1) an primären Zilien exprimiert wird. Erstaunlicherweise traf dies auch für fast alle in den Folgejahren identifizierten Genprodukte von Zystengenen zu, egal ob es sich hierbei um Gene handelte, deren Mutationen zu polyzystischen Nierenerkrankungen (ARPKD, ADPKD), zu einer Nephronophthise oder zum Bardet – Biedl Syndrom führten. Gleichzeitig konnte in umfangreichen funktionellen Untersuchungen gezeigt werden, dass Einschränkungen der ziliären Funktion an renalen Tubulusepithelien zur Ausbildung von Nierenzysten führen. Dies hatte zur Folge, dass die zystischen Nierenerkrankungen in der überwiegenden Mehrheit heute zur Erkrankungsgruppe der Zilienerkrankungen/ Ziliopathien gezählt werden. Vereinfachend könnte man sogar zusammenfassen: „Die Zilie macht die Zyste“. Allein, es fehlt weiterhin ein detailliertes Verständnis der beteiligten molekularbiologischen Prozesse. Deren Erforschung ist weiterhin Gegenstand umfangreicher wissenschaftlicher Bemühungen – nicht zuletzt von NEOCYST.